Oxidative addition
Oxidative addition and reductive elimination are two important and related classes of reactions in organometallic chemistry.[1][2][3][4] Oxidative addition is a process that increases both the oxidation state and coordination number of a metal centre. Oxidative addition is often a step in catalytic cycles, in conjunction with its reverse reaction, reductive elimination.[5]
Contents
1 Role in transition metal chemistry
2 Mechanisms of oxidative addition
2.1 Concerted pathway
2.2 SN2-type
2.3 Ionic
2.4 Radical
3 Applications
4 References
5 Further reading
6 External links
Role in transition metal chemistry
For transition metals, oxidative reaction results in the decrease in the dn to a configuration with fewer electrons, often 2e fewer. Oxidative addition is favored for metals that are (i) basic and/or (ii) easily oxidized. Metals with a relatively low oxidation state often satisfy one of these requirements, but even high oxidation state metals undergo oxidative addition, as illustrated by the oxidation of Pt(II) with chlorine:
- [PtCl4]2− + Cl2 → [PtCl6]2−
In classical organometallic chemistry, the formal oxidation state of the metal and the electron count of the complex both increase by two.[6] One-electron changes are also possible and in fact some oxidative addition reactions proceed via series of 1e changes. Although oxidative additions can occur with the insertion of a metal into many different substrates, oxidative additions are most commonly seen with H–H, H–X, and C–X bonds because these substrates are most relevant to commercial applications.
Oxidative addition requires that the metal complex have a vacant coordination site. For this reason, oxidative additions are common for four- and five-coordinate complexes.
Reductive elimination is the reverse of oxidative addition.[7] Reductive elimination is favored when the newly formed X–Y bond is strong. For reductive elimination to occur the two groups (X and Y) should be mutually adjacent on the metal's coordination sphere. Reductive elimination is the key product-releasing step of several reactions that form C–H and C–C bonds.[5]
Mechanisms of oxidative addition
Oxidative additions proceed via many pathways that depend on the metal center and the substrates.
Concerted pathway
Oxidative additions of nonpolar substrates such as hydrogen and hydrocarbons appear to proceed via concerted pathways. Such substrates lack π-bonds, consequently a three-centered σ complex is invoked, followed by intramolecular ligand bond cleavage of the ligand (probably by donation of electron pair into the sigma* orbital of the inter ligand bond) to form the oxidized complex. The resulting ligands will be mutually cis,[2] although subsequent isomerization may occur.

This mechanism applies to the addition of homonuclear diatomic molecules such as H2. Many C–H activation reactions also follow a concerted mechanism through the formation of an M–(C–H) agostic complex.[2]
A representative example is the reaction of hydrogen with Vaska's complex, trans-IrCl(CO)[P(C6H5)3]2. In this transformation, iridium changes its formal oxidation state from +1 to +3. The product is formally bound to three anions: one chloride and two hydride ligands. As shown below, the initial metal complex has 16 valence electrons and a coordination number of four whereas the product is a six-coordinate 18 electron complex.

Formation of a trigonal bipyramidal dihydrogen intermediate is followed by cleavage of the H–H bond, due to electron back donation into the H–H σ*-orbital. This system is also in chemical equilibrium, with the reverse reaction proceeding by the elimination of hydrogen gas with simultaneous reduction of the metal center.[8]
The electron back donation into the H–H σ*-orbital to cleave the H–H bond causes electron-rich metals to favor this reaction.[8] The concerted mechanism produces a cis dihydride, while the stereochemistry of the other oxidative addition pathways do not usually produce cis adducts.
SN2-type
Some oxidative additions proceed analogously to the well known bimolecular nucleophilic substitution reactions in organic chemistry. Nucleophillic attack by the metal center at the less electronegative atom in the substrate leads to cleavage of the R–X bond, to form an [M–R]+ species. This step is followed by rapid coordination of the anion to the cationic metal center. For example, reaction of a square planar complex with methyl iodide:
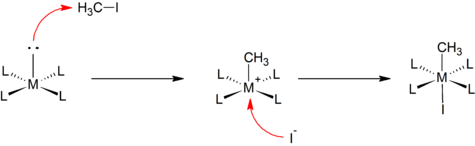
This mechanism is often assumed in the addition of polar and electrophilic substrates, such as alkyl halides and halogens.[2]
Ionic
The ionic mechanism of oxidative addition is similar to the SN2 type in that it involves the stepwise addition of two distinct ligand fragments. The key difference being that ionic mechanisms involve substrates which are dissociated in solution prior to any interactions with the metal center. An example of ionic oxidative addition is the addition of hydrochloric acid.[2]
Radical
In addition to undergoing SN2-type reactions, alkyl halides and similar substrates can add to a metal center via a radical mechanism, although some details remain controversial.[2] Reactions which are generally accepted to proceed by a radical mechanism are known however. One example was proposed by Lednor and co-workers.[9]
- Initiation
[(CH3)2C(CN)N]2 → 2 (CH3)2(CN)C• + N2
- (CH3)2(CN)C• + PhBr → (CH3)2(CN)CBr + Ph•
- Propagation
- Ph• + [Pt(PPh3)2] → [Pt(PPh3)2Ph]•
- [Pt(PPh3)2Ph]• + PhBr → [Pt(PPh3)2PhBr] + Ph•
Applications
Oxidative addition and reductive elimination are invoked in many catalytic processes both in homogeneous catalysis (i.e., in solution) such as the Monsanto process and alkene hydrogenation using Wilkinson's catalyst. It is often suggested that oxidative addition-like reactions are also involved in mechanisms of heterogeneous catalysis, e.g. hydrogenation catalyzed by platinum metal. Metals are however characterised by band structures, so oxidation states are not meaningful. Oxidative addition is also needed in order for nucleophilic addition of an alkyl group to occur. Oxidative insertion is also a crucial step in many cross-coupling reactions like the Suzuki coupling, Negishi coupling, and the Sonogashira coupling.
References
^ Jay A. Labinger "Tutorial on Oxidative Addition" Organometallics, 2015, volume 34, pp 4784–4795. doi:10.1021/acs.organomet.5b00565
^ abcdef Crabtree, Robert (2005). The Organometallic Chemistry of the Transition Metals. Wiley-Interscience. pp. 159–180. ISBN 0-471-66256-9..mw-parser-output cite.citation{font-style:inherit}.mw-parser-output .citation q{quotes:"""""""'""'"}.mw-parser-output .citation .cs1-lock-free a{background:url("//upload.wikimedia.org/wikipedia/commons/thumb/6/65/Lock-green.svg/9px-Lock-green.svg.png")no-repeat;background-position:right .1em center}.mw-parser-output .citation .cs1-lock-limited a,.mw-parser-output .citation .cs1-lock-registration a{background:url("//upload.wikimedia.org/wikipedia/commons/thumb/d/d6/Lock-gray-alt-2.svg/9px-Lock-gray-alt-2.svg.png")no-repeat;background-position:right .1em center}.mw-parser-output .citation .cs1-lock-subscription a{background:url("//upload.wikimedia.org/wikipedia/commons/thumb/a/aa/Lock-red-alt-2.svg/9px-Lock-red-alt-2.svg.png")no-repeat;background-position:right .1em center}.mw-parser-output .cs1-subscription,.mw-parser-output .cs1-registration{color:#555}.mw-parser-output .cs1-subscription span,.mw-parser-output .cs1-registration span{border-bottom:1px dotted;cursor:help}.mw-parser-output .cs1-ws-icon a{background:url("//upload.wikimedia.org/wikipedia/commons/thumb/4/4c/Wikisource-logo.svg/12px-Wikisource-logo.svg.png")no-repeat;background-position:right .1em center}.mw-parser-output code.cs1-code{color:inherit;background:inherit;border:inherit;padding:inherit}.mw-parser-output .cs1-hidden-error{display:none;font-size:100%}.mw-parser-output .cs1-visible-error{font-size:100%}.mw-parser-output .cs1-maint{display:none;color:#33aa33;margin-left:0.3em}.mw-parser-output .cs1-subscription,.mw-parser-output .cs1-registration,.mw-parser-output .cs1-format{font-size:95%}.mw-parser-output .cs1-kern-left,.mw-parser-output .cs1-kern-wl-left{padding-left:0.2em}.mw-parser-output .cs1-kern-right,.mw-parser-output .cs1-kern-wl-right{padding-right:0.2em}
^ Miessler, Gary L.; Tarr, Donald A. Inorganic Chemistry (3rd ed.).
[ISBN missing]
^ Shriver, D. F.; Atkins, P. W. Inorganic Chemistry.
[ISBN missing]
^ ab Hartwig, J. F. (2010). Organotransition Metal Chemistry, from Bonding to Catalysis. New York: University Science Books. ISBN 1-891389-53-X.
^ IUPAC, Compendium of Chemical Terminology, 2nd ed. (the "Gold Book") (1997). Online corrected version: (2006–) "oxidative addition". doi:10.1351/goldbook.O04367
^ IUPAC, Compendium of Chemical Terminology, 2nd ed. (the "Gold Book") (1997). Online corrected version: (2006–) "reductive elimination". doi:10.1351/goldbook.R05223
^ ab Johnson, Curtis; Eisenberg, Richard (1985). "Stereoselective Oxidative Addition of Hydrogen to Iridium(I) Complexes. Kinetic Control Based on Ligand Electronic Effects". Journal of the American Chemical Society. 107 (11): 3148–3160. doi:10.1021/ja00297a021.
^ Hall, Thomas L.; Lappert, Michael F.; Lednor, Peter W. (1980). "Mechanistic studies of some oxidative-addition reactions: free-radical pathways in the Pt0-RX, Pt0-PhBr, and PtII-R′SO2X Reactions (R = alkyl, R′ = aryl, X = halide) and in the related rhodium(I) or iridium(I) Systems". J. Chem. Soc., Dalton Trans. (8): 1448–1456. doi:10.1039/DT9800001448.
Further reading
Ananikov, Valentine P.; Musaev, Djamaladdin G.; Morokuma, Keiji (2005). "Theoretical Insight into the C−C Coupling Reactions of the Vinyl, Phenyl, Ethynyl, and Methyl Complexes of Palladium and Platinum". Organometallics. 24 (4): 715. doi:10.1021/om0490841.
External links
Toreki, R. "Oxidative Addition". The Organometallic HyperTextBook. Interactive Learning Paradigms Inc.
Toreki, R. "Reductive Elimination". The Organometallic HyperTextBook. Interactive Learning Paradigms Inc.


