Reductive elimination
Reductive elimination is an elementary step in organometallic chemistry in which the oxidation state of the metal center decreases while forming a new covalent bond between two ligands. It is the microscopic reverse of oxidative addition, and is often the product-forming step in many catalytic processes. Since oxidative addition and reductive elimination are reverse reactions, the same mechanisms apply for both processes, and the product equilibrium depends on the thermodynamics of both directions.[1][2]
Contents
1 General information
2 Mechanisms
2.1 Octahedral complexes
2.2 Square planar complexes
3 Factors that affect reductive elimination
3.1 Metal identity and electron density
3.2 Sterics
3.3 Participating ligands
3.4 Coordination number
3.5 Geometry
3.6 Photolysis/oxidation
4 Applications
5 References
General information
Reductive elimination is often seen in higher oxidation states, and can involve a two-electron change at a single metal center (mononuclear) or a one-electron change at each of two metal centers (binuclear, dinuclear, or bimetallic).[1][2]
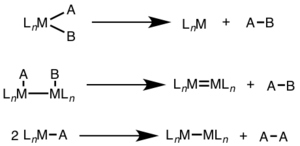
For mononuclear reductive elimination, the oxidation state of the metal decreases by two, while the d-electron count of the metal increases by two. This pathway is common for d8 metals Ni(II), Pd(II), and Au(III) and d6 metals Pt(IV), Pd(IV), Ir(III), and Rh(III). Additionally, mononuclear reductive elimination requires that the groups being eliminated must be cis to one another on the metal center.[3]

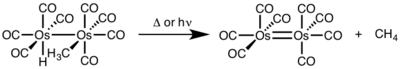
For binuclear reductive elimination, the oxidation state of each metal decreases by one, while the d-electron count of each metal increases by one. This type of reactivity is generally seen with first row metals, which prefer a one-unit change in oxidation state, but has been observed in both second and third row metals.[4]
Mechanisms
As with oxidative addition, several mechanisms are possible with reductive elimination. The prominent mechanism is a concerted pathway, meaning that it is a nonpolar, three-centered transition state with retention of stereochemistry. In addition, an SN2 mechanism, which proceeds with inversion of stereochemistry, or a radical mechanism, which proceeds with obliteration of stereochemistry, are other possible pathways for reductive elimination.[1]
Octahedral complexes
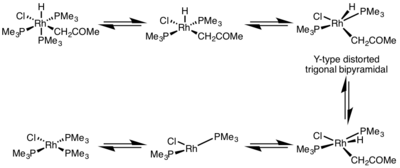
The rate of reductive elimination is greatly influenced by the geometry of the metal complex. In octahedral complexes, reductive elimination can be very slow from the coordinatively saturated center, and often, reductive elimination only proceeds via a dissociative mechanism, where a ligand must initially dissociate to make a five-coordinate complex. This complex adopts a Y-type distorted trigonal bipyramidal structure where a π-donor ligand is at the basal position and the two groups to be eliminated are brought very close together. After elimination, a T-shaped three-coordinate complex is formed, which will associate with a ligand to form the square planar four-coordinate complex.[5]
Square planar complexes
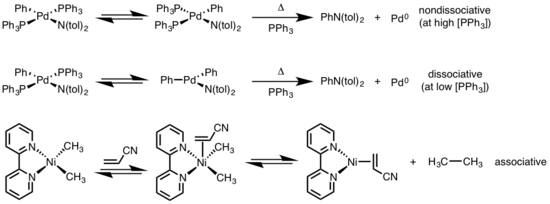
Reductive elimination of square planar complexes can progress through a variety of mechanisms: dissociative, nondissociative, and associative. Similar to octahedral complexes, a dissociative mechanism for square planar complexes initiates with loss of a ligand, generating a three-coordinate intermediate that undergoes reductive elimination to produce a one-coordinate metal complex. For a nondissociative pathway, reductive elimination occurs from the four-coordinate system to afford a two-coordinate complex. If the eliminating ligands are trans to each other, the complex must first undergo a trans to cis isomerization before eliminating. In an associative mechanism, a ligand must initially associate with the four-coordinate metal complex to generate a five-coordinate complex that undergoes reductive elimination synonymous to the dissociation mechanism for octahedral complexes.[6][7]
Factors that affect reductive elimination
Reductive elimination is sensitive to a variety of factors including: 1) metal identity and electron density; 2) sterics; 3) participating ligands; 4) coordination number; 5) geometry; and 6) photolysis/oxidation. Additionally, because reductive elimination and oxidative addition are reverse reactions, any sterics or electronics that enhance the rate of reductive elimination must thermodynamically hinder the rate of oxidative addition.[2]
Metal identity and electron density
First-row metal complexes tend to undergo reductive elimination faster than second-row metal complexes, which tend to be faster than third-row metal complexes. This is due to bond strength, with metal-ligand bonds in first-row complexes being weaker than metal-ligand bonds in third-row complexes. Additionally, electron-poor metal centers undergo reductive elimination faster than electron-rich metal centers since the resulting metal would gain electron density upon reductive elimination.[8]

Sterics
Reductive elimination generally occurs more rapidly from a more sterically hindered metal center because the steric encumbrance is alleviated upon reductive elimination. Additionally, wide ligand bite angles generally accelerate reductive elimination because the sterics force the eliminating groups closer together, which allows for more orbital overlap.[9]

Participating ligands
Kinetics for reductive elimination are hard to predict, but reactions that involve hydrides are particularly fast due to effects of orbital overlap in the transition state.[10]

Coordination number
Reductive elimination occurs more rapidly for complexes of three- or five-coordinate metal centers than for four- or six-coordinate metal centers. For even coordination number complexes, reductive elimination leads to an intermediate with a strongly metal-ligand antibonding orbital. When reductive elimination occurs from odd coordination number complexes, the resulting intermediate occupies a nonbonding molecular orbital.[11]

Geometry
Reductive elimination generally occurs faster for complexes whose structures resemble the product.[2]
Photolysis/oxidation
Reductive elimination can be induced by oxidizing the metal center to a higher oxidation state via light or an oxidant.[12]
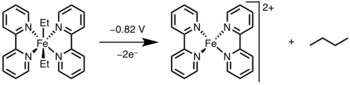
Applications
Reductive elimination has found widespread application in academia and industry, most notable being hydrogenation,[13] the Monsanto acetic acid process,[14]hydroformylation,[15] and cross coupling reactions.[16] In many of these catalytic cycles, reductive elimination is the product forming step and regenerates the catalyst; however, in the Heck reaction[17] and Wacker process,[18] reductive elimination is involved only in catalyst regeneration, as the products in these reactions are formed via β–hydride elimination.
References
^ abc Crabtree, Robert H. (2014). The Organometallic Chemistry of the Transition Metals (6 ed.). Wiley. p. 173. ISBN 978-1-118-13807-6..mw-parser-output cite.citation{font-style:inherit}.mw-parser-output .citation q{quotes:"""""""'""'"}.mw-parser-output .citation .cs1-lock-free a{background:url("//upload.wikimedia.org/wikipedia/commons/thumb/6/65/Lock-green.svg/9px-Lock-green.svg.png")no-repeat;background-position:right .1em center}.mw-parser-output .citation .cs1-lock-limited a,.mw-parser-output .citation .cs1-lock-registration a{background:url("//upload.wikimedia.org/wikipedia/commons/thumb/d/d6/Lock-gray-alt-2.svg/9px-Lock-gray-alt-2.svg.png")no-repeat;background-position:right .1em center}.mw-parser-output .citation .cs1-lock-subscription a{background:url("//upload.wikimedia.org/wikipedia/commons/thumb/a/aa/Lock-red-alt-2.svg/9px-Lock-red-alt-2.svg.png")no-repeat;background-position:right .1em center}.mw-parser-output .cs1-subscription,.mw-parser-output .cs1-registration{color:#555}.mw-parser-output .cs1-subscription span,.mw-parser-output .cs1-registration span{border-bottom:1px dotted;cursor:help}.mw-parser-output .cs1-ws-icon a{background:url("//upload.wikimedia.org/wikipedia/commons/thumb/4/4c/Wikisource-logo.svg/12px-Wikisource-logo.svg.png")no-repeat;background-position:right .1em center}.mw-parser-output code.cs1-code{color:inherit;background:inherit;border:inherit;padding:inherit}.mw-parser-output .cs1-hidden-error{display:none;font-size:100%}.mw-parser-output .cs1-visible-error{font-size:100%}.mw-parser-output .cs1-maint{display:none;color:#33aa33;margin-left:0.3em}.mw-parser-output .cs1-subscription,.mw-parser-output .cs1-registration,.mw-parser-output .cs1-format{font-size:95%}.mw-parser-output .cs1-kern-left,.mw-parser-output .cs1-kern-wl-left{padding-left:0.2em}.mw-parser-output .cs1-kern-right,.mw-parser-output .cs1-kern-wl-right{padding-right:0.2em}
^ abcd Hartwig, John F. (2010). Organotransition Metal Chemistry, from Bonding to Catalysis. University Science Books. p. 321. ISBN 978-1-891389-53-5.
^ Gillie, A.; Stille, J. K. (1980). "Mechanisms of 1,1-Reductive Elimination from Palladium". J. Am. Chem. Soc. 102: 4933. doi:10.1021/ja00535a018.
^ Okrasinski, S. J.; Nortom, J. R. (1977). "Mechanism of Reductive Elimination. 2. Control of Dinuclear vs. Mononuclear Elimination of Methane from cis-Hydridomethyltetracarbonylosmium". J. Am. Chem. Soc. 99: 295. doi:10.1021/ja00443a076.
^ Milstein, D. (1982). "The First Isolated, Stable cis-Hydridoalkylrhodium Complexes and Their reductive Elimination Reaction". J. Am. Chem. Soc. 104: 5227. doi:10.1021/ja00383a039.
^ Driver, M. S.; Hartwig, J. F. (1997). "Carbon−Nitrogen-Bond-Forming Reductive Elimination of Arylamines from Palladium(II) Phosphine Complexes". J. Am. Chem. Soc. 119: 8232. doi:10.1021/ja971057x.
^ Yamamoto, T.; Yamamoto, A.; Ikeda, S. (1971). "Study of Organo(dipyridyl)nickel Complexes. I. Stability and Activation of the Alkyl-Nickel Bonds of Dialkyl(dipyridyl)nickel by Coordination with Various Substituted Olefins". J. Am. Chem. Soc. 93: 3350. doi:10.1021/ja00743a009.
^ Giovannini, R.; Stüdemann, T.; Dussin, G.; Knochel, P. (1998). "An Efficient Nickel-Catalyzed Cross-Coupling Between sp3 Carbon Centers". Angew. Chem. Int. Ed. 37: 2387. doi:10.1002/(SICI)1521-3773(19980918)37:17<2387::AID-ANIE2387>3.0.CO;2-M.
^ Marcone, J. E.; Moloy, K. G. (1998). "Kinetic Study of Reductive Elimination from the Complexes (Diphosphine)Pd(R)(CN)". J. Am. Chem. Soc. 120: 8527. doi:10.1021/ja980762i.
^ Low, J. J.; Goddard, III, W. A. (1984). "Reductive Coupling of Hydrogen-Hydrogen, Hydrogen-Carbon, and Carbon-Carbon Bonds from Palladium Complexes". J. Am. Chem. Soc. 106: 8321. doi:10.1021/ja00338a067.
^ Crumpton-Bregel, D. M.; Goldberg, K. I. (2003). "Mechanisms of C-C and C-H Alkane Reductive Eliminations from Octahedral Pt(IV): Reaction via Five-Coordinate Intermediates or Direct Elimination?". J. Am. Chem. Soc. 125: 9442. doi:10.1021/ja029140u.
^ Lau, W.; Huffman, J. C.; Kochi, J. K. (1982). "Electrochemical Oxidation-Reduction of Organometallic Complexes. Effect of the Oxidation State on the Pathways for Reductive Elimination of Dialkyliron Complexes". Organometallics. 1: 155. doi:10.1021/om00061a027.
^ de Vries, J. G. (2007). The Handbook of Homogeneous Hydrogenation. Wiley. ISBN 978-3-527-31161-3.
^ Paulik, F. E.; Roth, J. F. (1968). "Novel Catalysts for the Low-pressure Carbonylation of Methanol to Acetic Acid". Chem. Commun.: 1578. doi:10.1039/C1968001578A.
^ Ojima, I.; Tsai, C.-H.; Tzamarioudaki, M.; Bonafoux, D. (2004). "The Hydroformylation Reaction". Organic Reactions. 56: 1. doi:10.1002/0471264180.or056.01.
^ New Trends in Cross-Coupling: Theory and Applications Thomas Colacot (Editor) 2014
ISBN 978-1-84973-896-5
^ de Vries, J. G. (2001). "The Heck reaction in the production of fine chemicals". Can. J. Chem. 79: 1086. doi:10.1139/v01-033.
^ Dong, J. J.; Browne, W. R.; Feringa, B. L. (2015). "Palladium-Catalyzed anti-Markovnikov Oxidation of Terminal Alkenes". Angew. Chem. Int. Ed. 54: 734. doi:10.1002/anie.201404856.


