DNA測序
| 本条目需要擴充。(2013年5月26日) |
DNA测序(DNA sequencing,或譯DNA定序)是指分析特定DNA片段的碱基序列,也就是腺嘌呤(A)、胸腺嘧啶(T)、胞嘧啶(C)與鳥嘌呤的(G)排列方式。快速的DNA测序方法的出现极大地推动了生物学和医学的研究和发现。
在基础生物学研究中,和在众多的应用领域,如诊断,生物技术,法医生物学,生物系统学中,DNA序列知识已成为不可缺少的知识。具有现代的DNA测序技术的快速测序速度已经有助于达到测序完整的DNA序列,或多种类型的基因组测序和生命物种,包括人类基因组和其他许多动物,植物和微生物物种的完整DNA序列。
RNA測序則通常将RNA提取后,反转录为DNA后使用DNA测序的方法进行测序。目前应用最广泛的是由弗雷德里克·桑格发明的Sanger双脱氧链终止法(Chain Termination Method)[1]。新的测序方法,例如454生物科学的方法和焦磷酸测序法。
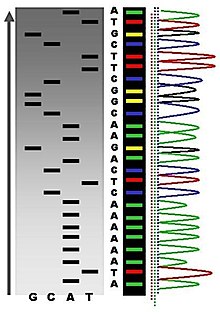
自动化chain-termination DNA测序结果的一个例子.
目录
1 应用
1.1 分子生物学
1.2 进化生物学
1.3 宏基因组学(或元基因组学)
1.4 医学
1.5 法医学
2 历史
2.1 DNA结构与功能的发现
2.2 RNA测序
2.3 全基因组测序
3 基本方法
3.1 Maxam-Gilbert测序法
3.2 Sanger测序法
4 高级方法和de novo测序法
4.1 霰彈槍定序法
4.2 Bridge PCR
5 新一代测序
5.1 454生物科学和焦磷酸测序法
6 正在开发的测序法
6.1 纳米孔DNA测序法
7 參見
8 参考文献
应用
DNA测序可用于确定任何生物的单个基因的序列,较大的遗传区域(即基因簇或操纵子的簇),完整的染色体或整个基因组。 DNA测序也是对RNA或蛋白质进行测序的最有效方法(通过对开放阅读框测序)。目前,DNA测序已成为生物学和其他科学领域(如医学,法医学或人类学等)的关键技术。
分子生物学
在分子生物学中,DNA测序可被用于研究基因组及其编码的蛋白质。利用测序获得的信息,科研人员能够识别基因的变化,基因与疾病和表型的关联,并确定潜在的药物靶点。
进化生物学
由于DNA是携带有遗传信息的大分子,在进化生物学中,DNA测序被用于研究不同生物体之间的相关性以及它们是如何进化的。
宏基因组学(或元基因组学)
宏基因组学是一门直接取得环境中所有遗传物质的研究。环境包括但不限于水体,污水,污垢,从空气中过滤出的碎片或者从生物体采集的样本。了解在特定环境中存在哪些生物体对于生态学,流行病学,微生物学和其他领域的研究至关重要。DNA测序使研究人员能够确定微生物群中可能存在哪些类型的微生物。
医学
医疗人员可通过对患者基因(基因组)的测序结果确定该患者是否有携带遗传性疾病的风险。需要注意的是,该方法属于基因检测,有些基因检测不会用到DNA测序技术。
法医学
DNA测序可以与DNA图谱鉴定(基因指纹分析,英语:DNA profiling)一起用于法医鉴定和亲子鉴定。 DNA测试在过去的几十年中发展迅猛,目前已能够做到将DNA鉴定结果与被调查对象联系起来。指纹,唾液,毛囊等中的DNA特征可以将不同的生物体进行区分。测试DNA是一种可以检测DNA链中特定基因组并生成唯一的个性化DNA模型的技术。每一种有机体都有其DNA特征,并可以通过DNA测试来确定。两个人具有完全相同的DNA特征是非常罕见的,因此保证了DNA测试的成功。
历史
DNA结构与功能的发现

弗雷德里克·桑格,DNA测序的先驱者。桑格是少数获得两项诺贝尔奖的科学家之一,其中一项为蛋白质测序,另一项为DNA测序。
脱氧核糖核酸(DNA)最早在1869年由Friedrich Miescher发现并分离出来,但由于当时普遍认为遗传信息保存于蛋白质而不是DNA中,因此在过去几十年中DNA一直没有得到充分研究。1944年,由于Oswald Avery,Colin MacLeod和Maclyn McCarty的一些实验表明,纯化的DNA可以将一种细菌变成另一种细菌,这种情况才发生了变化。这也是首次DNA显示出改变细胞特性的能力。
1953年,James Watson和Francis Crick根据Rosalind Franklin研究的结晶X射线结构提出了他们的双螺旋DNA模型。根据该模型,DNA由彼此缠绕的两条核苷酸链组成,通过氢键连接在一起并以相反方向运行。每条链由四个互补的核苷酸组成:腺嘌呤(A),胞嘧啶(C),鸟嘌呤(G)和胸腺嘧啶(T),其中A与T配对,C与G配对。他们提出的这种结构,使得每条单链都可被用于重建另一条链,并且让遗传信息代代相传。
对蛋白质进行测序的基础首先由弗雷德里克·桑格(Frederick Sanger)的工作奠定,他于1955年完成了胰岛素(胰腺分泌的一种蛋白质)中所有氨基酸序列的测序工作。这是首个确凿的证据证明蛋白质是具有特定分子模式的化学实体,而不是悬浮在流体中的随机混合物。桑格在胰岛素测序方面的成功使得X射线晶体学家大为振奋,包括沃森和克里克,他们现在正试图理解DNA如何指导细胞内蛋白质的形成。在1954年10月弗雷德里克·桑格出席一系列讲座后不久,克里克开始发展一种理论,认为DNA中核苷酸的排列决定了蛋白质中氨基酸的序列,从而帮助确定蛋白质的功能。他于1958年发表了这一理论。
RNA测序
RNA测序是最早的核苷酸测序形式之一。 RNA测序的主要标志是1972年和1976年Walter Fiers及其同事在根特大学(根特,比利时)确定并发表的第一个完整基因序列和噬菌体MS2的完整基因组。传统的RNA测序方法需要创建一个用于测序的互补cDNA(Complementary DNA)分子。
全基因组测序
第一个完整的DNA基因组测序是在1977年噬菌体φX174的测序工作。医学研究委员会的科学家在1984年破译了Epstein-Barr病毒的完整DNA序列,发现它含有172,282个核苷酸。 该序列的完成标志着DNA测序的一个重要转折点,它在没有病毒基因谱知识的情况下实现了DNA测序。
20世纪80年代初,Pohl及其同事开发了一种在电泳时将测序反应混合物的DNA分子转移到固定基质上的非放射性方法。随后GATC Biotech公司的DNA测序仪“Direct-Blotting-Electrophoresis-System GATC 1500”商业化,该测序仪在EU基因组测序程序的框架以及酵母酿酒酵母染色体II的完整DNA序列中广泛使用。加利福尼亚理工学院的Leroy E. Hood实验室于1986年宣布了第一台半自动DNA测序机。随后,Applied Biosystems在1987年推出了第一台全自动测序仪ABI 370。以及Dupont公司的Genesis 2000,该仪器使用了一种新的荧光标记技术,可在单一泳道中识别所有四个双脱氧核苷酸。到1990年,美国国立卫生研究院(NIH)已开始对支原体,大肠杆菌,秀丽隐杆线虫和酿酒酵母进行大规模测序实验,费用为每个碱基0.75美元。同时,人类cDNA序列的测序始于Craig Venter的实验室,试图获取人类基因组的编码部分。 1995年,Venter,Hamilton Smith及其基因组研究所(TIGR)的同事发表了第一个完整的自由生物体细菌流感嗜血杆菌(Haemophilus influenzae)的基因组。该环形染色体中含有1,830,137个碱基,其在《科学》杂志中的发表标志着全基因组鸟枪法测序的首次公开使用,摆脱了初始绘制工作的需要。
基本方法
Maxam-Gilbert测序法
马克萨姆-吉尔伯特测序(英语:Maxam-Gilbert sequencing)是一项由阿伦·马克萨姆与沃尔特·吉尔伯特于1976~1977年间开发的DNA测序方法。此项方法基于:对核鹼基特异性地进行局部化学改性,接下来在改性核苷酸毗邻的位点处DNA骨架发生断裂[2] 。
Sanger测序法
Sanger(桑格)双脱氧链终止法是弗雷德里克·桑格(Frederick Sanger)于1975年发明的。测序过程需要先做一个聚合酶连锁反应(PCR)。PCR过程中,双脱氧核苷酸可能随机地被加入到正在合成中的DNA片段里。由于双脱氧核糖核苷酸又少了一个氧原子,一旦它被加入到DNA链上,这个DNA链就不能继续增加长度。最终的结果是获得所有可能获得的、不同长度的DNA片段。目前最普遍最先进的方法,是将双脱氧核糖核苷酸进行不同荧光标记。将PCR反应获得的总DNA通过毛细管电泳分离,跑到最末端的DNA就可以在激光的作用下发出荧光。由于ddATP, ddGTP, ddCTP, ddTTP(4种双脱氧核糖核苷酸)荧光标记不同,计算机可以自动根据颜色判断该位置上碱基究竟是A,T,G,C中的哪一个[3]。
高级方法和de novo测序法
霰彈槍定序法
霰彈槍定序法(Shotgun sequencing,又称鸟枪法)是一种广泛使用的为长DNA测序的方法,比傳統的定序法快速,但精確度較差。曾經使用於塞雷拉基因組(Celera Genomics)公司所主持的人類基因組计划。
Bridge PCR
新一代测序
随着人们对低成本测序的需求与日俱增,推动了高通量测序(或称为二代测序、新一代测序、下一代测序)的发展,这些技术对测序过程多路复用,同时产生上千或上百万条序列[4][5]。高通量测序技术的目的是降低DNA测序的成本,这个成本比同样可实现测序的染料终止法来得低得多[6]。超高通量测序过程中可同时运行高达500,000次的边合成边测序[7][8][9]。

需要根据多个片段序列所重叠的区域将它们全部组装起来。
| 方法 | 单分子实时测序(Pacific Bio) | 离子半导体(Ion Torrent sequencing) | 焦磷酸测序(454) | 边合成边测序(Illumina) | 边连接边测序(SOLiD sequencing) | 链终止法(Sanger sequencing) |
|---|---|---|---|---|---|---|
| 读长 | 5,500 bp to 8,500 bp avg (10,000 bp N50); maximum read length >30,000 bases[12][13][14] | up to 400 bp | 700 bp | 50 to 300 bp | 50+35 or 50+50 bp | 400 to 900 bp |
| 精确度 | 99.999% consensus accuracy; 87% single-read accuracy[15] | 98% | 99.9% | 98% | 99.9% | 99.9% |
| 每次运行可获取读段数 | 50,000 per SMRT cell, or ~400 megabases[16][17] | up to 80 million | 1 million | up to 3 billion | 1.2 to 1.4 billion | N/A |
| 每次运行耗时 | 30 minutes to 2 hours [18] | 2 hours | 24 hours | 1 to 10 days, depending upon sequencer and specified read length[19] | 1 to 2 weeks | 20 minutes to 3 hours |
| 每百万碱基所耗成本(美元) | $0.33-$1.00 | $1 | $10 | $0.05 to $0.15 | $0.13 | $2400 |
| 优势 | Longest read length. Fast. Detects 4mC, 5mC, 6mA.[20] | Less expensive equipment. Fast. | Long read size. Fast. | Potential for high sequence yield, depending upon sequencer model and desired application. | Low cost per base. | Long individual reads. Useful for many applications. |
| 劣势 | Moderate throughput. Equipment can be very expensive. | Homopolymer errors. | Runs are expensive. Homopolymer errors. | Equipment can be very expensive. Requires high concentrations of DNA. | Slower than other methods. Have issue sequencing palindromic sequence.[21] | More expensive and impractical for larger sequencing projects. |
454生物科学和焦磷酸测序法
454测序法由454生物科学发明,是一个类似焦磷酸测序法的新方法。2003年向GenBank提交了一个腺病毒全序列[22],使得他们的技术成为Sanger测序法后第一个被用来测生物基因组全序列的新方法。454使用类似于焦磷酸测序的方法,有着相当高的读取速度,大约为5小时可以测两千万碱基对[23]。
正在开发的测序法
纳米孔DNA测序法
參見
- 蛋白質定序
- 已測序的生物
参考文献
^ http://www.bioon.com/experiment/nua2/89939.shtml
^ Maxam AM, Gilbert W. A new method for sequencing DNA. Proc. Natl. Acad. Sci. U.S.A. February 1977, 74 (2): 560–4. Bibcode:1977PNAS...74..560M. PMC 392330. PMID 265521. doi:10.1073/pnas.74.2.560.
^ http://en.wikipedia.org/wiki/Chain_termination_method
^ Hall, Nell. Advanced sequencing technologies and their wider impact in microbiology. J. Exp. Biol. May 2007, 209 (Pt 9): 1518–1525. PMID 17449817. doi:10.1242/jeb.001370.
^ Church, George M. Genomes for all. Sci. Am. January 2006, 294 (1): 46–54. PMID 16468433. doi:10.1038/scientificamerican0106-46.
^ 引用错误:没有为名为pmid18165802的参考文献提供内容
^ Kalb, Gilbert; Moxley, Robert. Massively Parallel, Optical, and Neural Computing in the United States. IOS Press. 1992. ISBN 90-5199-097-9. [页码请求]
^ PMID 18832462(PubMed)
本引用來源將會在數十分鐘後自動完成。您可以检查英文对应模板或手動擴充
^ PMID 19679224(PubMed)
本引用來源將會在數十分鐘後自動完成。您可以检查英文对应模板或手動擴充
^ Quail, Michael; Smith, Miriam E; Coupland, Paul; 等. A tale of three next generation sequencing platforms: comparison of Ion torrent, pacific biosciences and illumina MiSeq sequencers. BMC Genomics. 1 January 2012, 13 (1): 341. PMC 3431227. PMID 22827831. doi:10.1186/1471-2164-13-341.
^ Liu, Lin; Li, Yinhu; Li, Siliang; 等. Comparison of Next-Generation Sequencing Systems. Journal of Biomedicine and Biotechnology (Hindawi Publishing Corporation). 1 January 2012, 2012: 1–11. doi:10.1155/2012/251364.
^ New Products: PacBio's RS II; Cufflinks | In Sequence | Sequencing | GenomeWeb
^ After a Year of Testing, Two Early PacBio Customers Expect More Routine Use of RS Sequencer in 2012. GenomeWeb. 10 January 2012.
^ Pacific Biosciences Introduces New Chemistry With Longer Read Lengths
^ http://www.nature.com/nmeth/journal/v10/n6/full/nmeth.2474.html
^ De novo bacterial genome assembly: a solved problem? | In between lines of code
^ Rasko, David A.; Webster, Dale R.; Sahl, Jason W.; 等. Origins of the Strain Causing an Outbreak of Hemolytic–Uremic Syndrome in Germany. N Engl J Med. 25 August 2011, 365 (8): 709–717. doi:10.1056/NEJMoa1106920.
^ Tran, Ben; Brown, Andrew M.K.; Bedard, Philippe L.; Winquist, Eric; Goss, Glenwood D.; Hotte, Sebastien J.; Welch, Stephen A.; Hirte, Hal W.; Zhang, Tong; Stein, Lincoln D.; Ferretti, Vincent; Watt, Stuart; Jiao, Wei; Ng, Karen; Ghai, Sangeet; Shaw, Patricia; Petrocelli, Teresa; Hudson, Thomas J.; Neel, Benjamin G.; 等. Feasibility of real time next generation sequencing of cancer genes linked to drug response: Results from a clinical trial. Int. J. Cancer. 1 January 2012: 1547–1555. doi:10.1002/ijc.27817.
^ van Vliet, Arnoud H.M. Next generation sequencing of microbial transcriptomes: challenges and opportunities. FEMS Microbiology Letters. 1 January 2010, 302 (1): 1–7. doi:10.1111/j.1574-6968.2009.01767.x.
^ Murray, I. A.; Clark, T. A.; Morgan, R. D.; Boitano, M.; Anton, B. P.; Luong, K.; Fomenkov, A.; Turner, S. W.; Korlach, J.; Roberts, R. J. The methylomes of six bacteria. Nucleic Acids Research. 2 October 2012, 40 (22): 11450–62. PMC 3526280. PMID 23034806. doi:10.1093/nar/gks891. 引文使用过时参数coauthors (帮助)
^ Yu-Feng Huang, Sheng-Chung Chen, Yih-Shien Chiang, Tzu-Han Chen & Kuo-Ping Chiu. Palindromic sequence impedes sequencing-by-ligation mechanism. BMC systems biology. 2012,. 6 Suppl 2: S10. PMID 23281822. doi:10.1186/1752-0509-6-S2-S10.
^ 存档副本. [2006-11-17]. (原始内容存档于2006-10-29).
^ 存档副本. [2006-11-17]. (原始内容存档于2006-10-29).
| ||||||||||||||||||||||||

| ||||||||||||||||||||||


